nextflow
Nextflow workflow syntax highlighting and snippets for Sublime Text 4
Details
Installs
- Total 730
- Win 182
- Mac 371
- Linux 177
| Aug 6 | Aug 5 | Aug 4 | Aug 3 | Aug 2 | Aug 1 | Jul 31 | Jul 30 | Jul 29 | Jul 28 | Jul 27 | Jul 26 | Jul 25 | Jul 24 | Jul 23 | Jul 22 | Jul 21 | Jul 20 | Jul 19 | Jul 18 | Jul 17 | Jul 16 | Jul 15 | Jul 14 | Jul 13 | Jul 12 | Jul 11 | Jul 10 | Jul 9 | Jul 8 | Jul 7 | Jul 6 | Jul 5 | Jul 4 | Jul 3 | Jul 2 | Jul 1 | Jun 30 | Jun 29 | Jun 28 | Jun 27 | Jun 26 | Jun 25 | Jun 24 | Jun 23 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Windows | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mac | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Linux | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Readme
- Source
- raw.githubusercontent.com

Nextflow and nf-core workflow completions, commands, syntax highlighting and snippets for Sublime Text 4
- ⚠️WARNING⚠️: Sublime Text 3 is no longer supported by this package as of version 1.0.0. Only Sublime Text 4 is supported since this package requires Python 3.8 and new features in ST4.
- ❗NOTE❗: Only DSL-2 Nextflow workflows are supported by this package.
This package provides Nextflow workflow language:
- completions (
params.,conda,<PROCESS/WORKFLOW>.out.<emit name>) - informative popups for processes and sub-workflows
- commands (insert container directive, insert module import statement)
- syntax highlighting
- snippets
Essentially, this package tries to make Nextflow workflow development a bit easier especially when trying to develop nf-core conventions and best practices for developing Nextflow pipelines.
Nextflow completions and commands
Process module include command
ctrl+l,pin an.nffile where you want to import your process
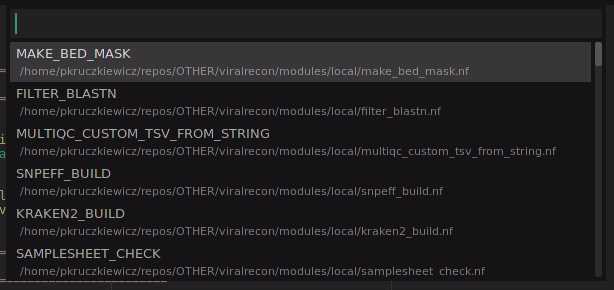
- select the process you wish to import
- something like the following will be inserted
include { MAKE_BED_MASK } from '../modules/local/make_bed_mask' addParams( options: modules['make_bed_mask'] )
The addParams( options: modules['make_bed_mask'] ) may not be needed and can be removed; it assumes that you have a conf/modules.config with a map of your module args, publish_dir, etc (see nf-core/modules for more info).
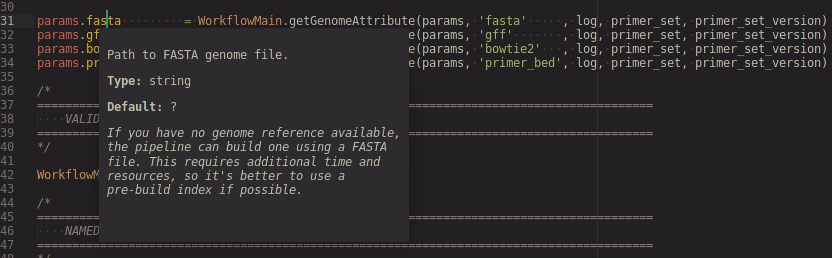
Workflow params
NOTE: Completions and info popups for params depend on a valid nextflow_schema.json in your workflow root directory. Example nextflow_schema.json for nf-core/viralrecon workflow.
Navigate cursor to a params.<variable> to show a popup with info pulled from the nextflow_schema.json for that workflow parameter.
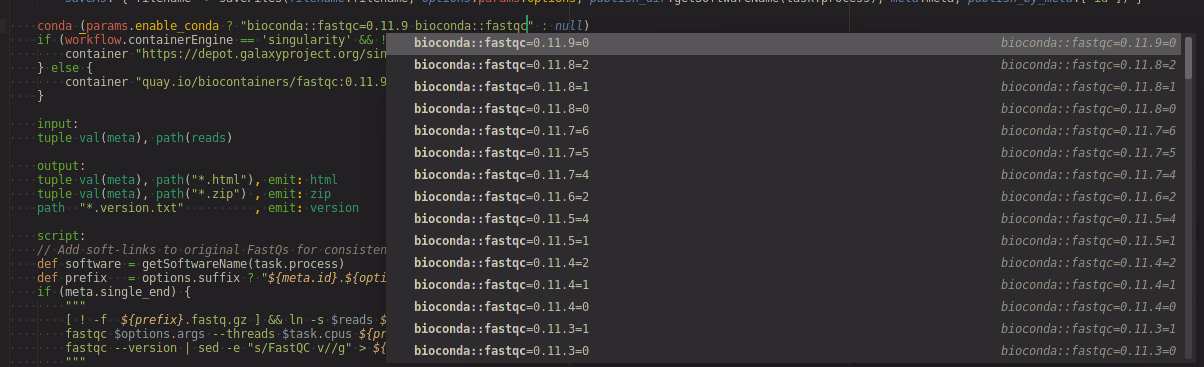
Conda completion
NOTE: Conda must be installed along with any channels (e.g. bioconda, conda-forge) to get packages information (needs to be able to run conda search).
- Open the command palette (
ctrl+shift+p) and run theNextflow: Fetch Conda packages informationcommand to fetch the latest Conda package info (runsconda search; may take a while). - In your process definition, inside the
condadirective string pressctrl+spaceto bring up the completion list. This may have a little delay since the package list may be very large.
process PANGOLIN {
conda '<press ctrl+space to bring up completion list>'
}
Process output channel completion
Get process named output (i.e. using the emit option) completions after typing <PROCESS_NAME>.out..
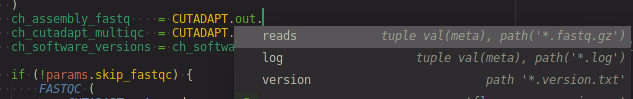
- completion shows what's on the channel as well!
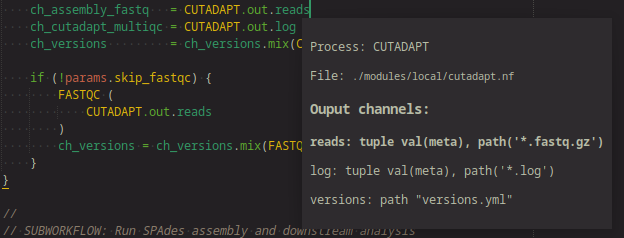
Process output channel popup
Show useful info about what output the a process is emitting.
Container directive insert command
This command inserts similar code to what you'd find in an nf-core modules process definition with respect to process container directives. The Biocontainers information is pulled from the Singularity images https://depot.galaxyproject.org/singularity/ and cached as a Python pickle file. Docker container image tags point to the Biocontainers Quay.io page.
- Open the command palette (
ctrl+shift+p) and run theNextflow: Fetch Biocontainers informationcommand to fetch the latest Biocontainers list fetched from - In your process definition, press
ctrl+l,c, search for the container you're interested in
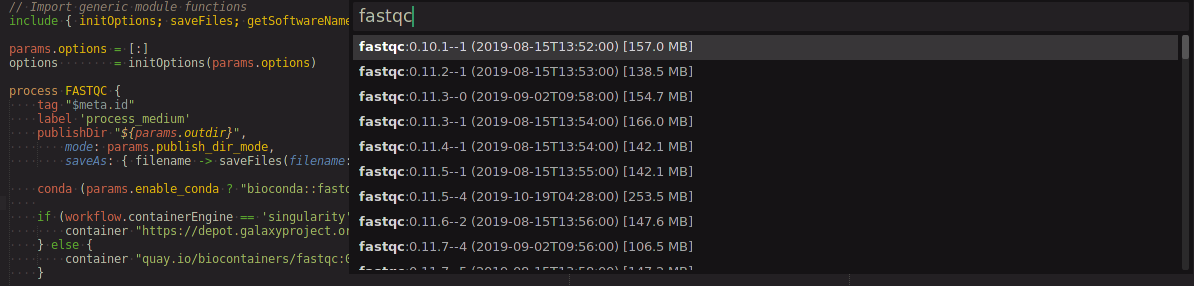
- Select the program and version you're interested in to output the following:
if (workflow.containerEngine == 'singularity' && !params.singularity_pull_docker_container) {
container 'https://depot.galaxyproject.org/singularity/fastqc:0.11.9--hdfd78af_1'
} else {
container 'quay.io/biocontainers/fastqc:0.11.9--hdfd78af_1'
}
Nextflow Syntax Highlighting
Nextflow syntax highlighting extends Sublime Text 4's Groovy syntax with highlighting of:
- imports (DSL-2 modules)
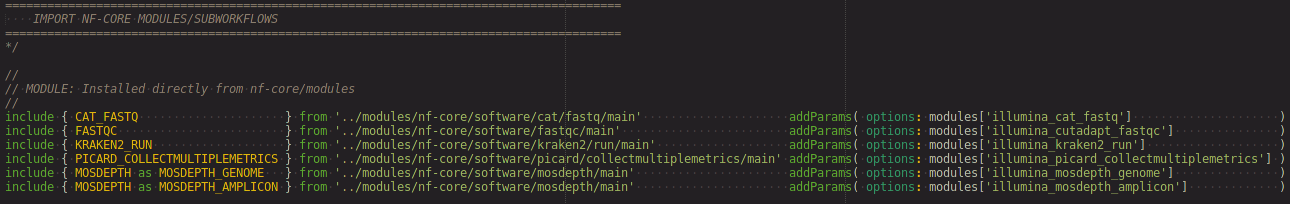
- workflow definitions
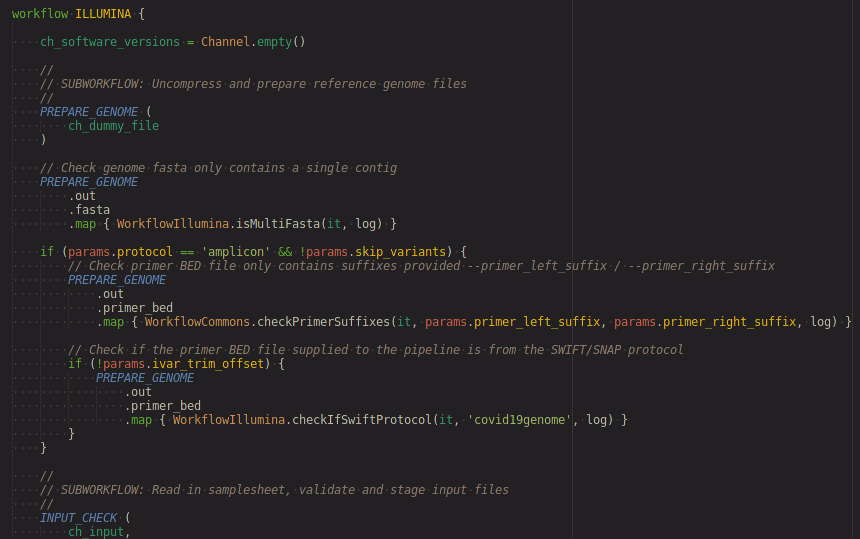
- process definitions
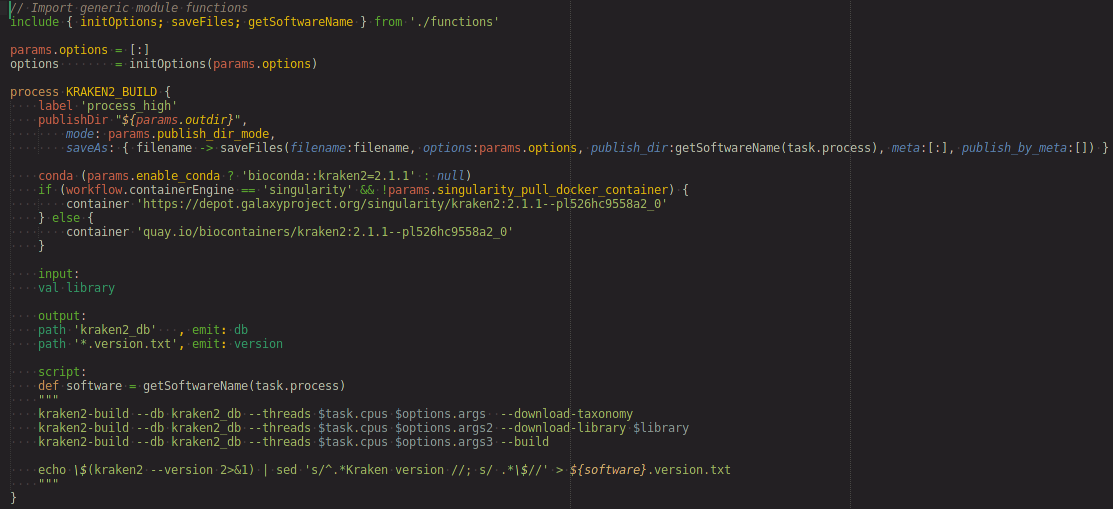
- channel highlighting based on matching
ch_* - some invalid syntax checks (into channel in input tag and from channel in output tag)
- highlighting special Nextflow functions and variables (
workflow,params,task, etc)
Process syntax highlighting and scoping allows one to easily go to the definition or usages of a process (ctrl+shift+g keyboard shortcut):
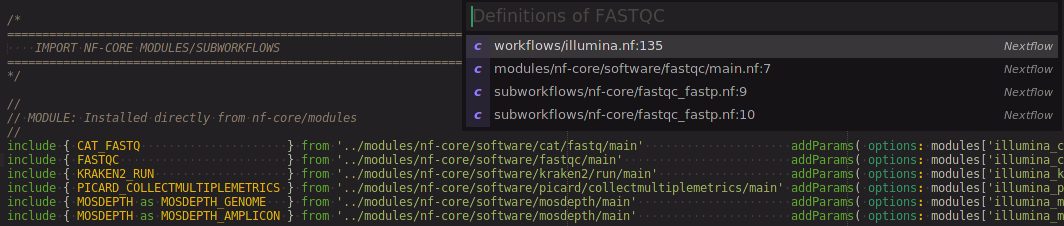
Nextflow Snippets
Type one of the following and press TAB:
!env:#!/usr/bin/env nextflowproc: process snippettag: tag process directive snippetpub: publishDir process directive snippetconda: conda process directive snippetillumina: Illumina paired-end reads file pairs channelcpus: insert"${task.cpus}"into a process scriptscript_path: specify user script (e.g. Python script) to use fromscripts/directory in workflow base directoryinfo:log.infosnippetdone: on workflow complete or error message
Changelog
1.1.0 - [2022-05-13]
Added:
- more informative popups for showing info about process output so it's easier to select the correct output channel without referencing the process code.
- subworkflow completions and info popups about
takeandemitchannels condadirective snippet
Fixed:
- comment toggling
1.0.0 - [2021-06-30]
- Added syntax highlighting for Nextflow DSL-2
- Added completions and commands for workflow
params,PROCESS.out., conda, container, module include - Removed some not very useful snippets
0.1.0-alpha.1 - [2019-03-27]
- Initial release with Groovy-based syntax highlighting and basic snippets
License
MIT License
Copyright © Peter Kruczkiewicz